| Journal of Food Bioactives, ISSN 2637-8752 print, 2637-8779 online |
| Journal website www.isnff-jfb.com |
Original Research
Volume 24, December 2023, pages 55-62
Pomelo peel extract induces ROS-mediated DNA double stand break and apoptotic death pathway in Caco-2 colon cancer cells
Vo Thi Hong Thama, Hsiao-Chi Wangb, #, Yung-Lin Chua, *, #
aDepartment of Food Science, College of Agriculture, National Pingtung University of Science and Technology, Pingtung, Taiwan
bDepartment of Cosmetics Applications and Management, Cardinal Tien Junior College of Healthcare and Management, No. 112, Minzu Road, Sindian District, New Taipei, Taiwan
#These authors contributed equally to this work.
*Corresponding author: Yung-Lin Chu, Department of Food Science, College of Agriculture, National Pingtung University of Science and Technology, Pingtung, Taiwan. Tel: +886-8-7703202 # 7007; E-mail: ylchu@mail.npust.edu.tw
DOI: 10.31665/JFB.2023.18364
Received: December 25, 2023
Revised received & accepted: December 29, 2023
| Abstract | ▴Top |
Colorectal cancer (CRC) ranks among the top ten leading causes of death in the world. Beyond chemical therapy or targeted therapy, finding an alternative approach to colorectal cancer therapy is necessary. Citrus grandis (L.) Osbeck is a popular fruit cultivated worldwide; the peel is mainly discarded after consuming the fruit. However, it has also been commonly reported as folk medicine for treating several illnesses, such as cough and indigestion. In this study, Caco-2 cells were treated with different concentrations of pomelo peel extract (PPE). PPE suppressed cell viability in a dose-dependent manner at 24 and 48 h. The PPE extract showed condensed chromatin and apoptotic cells at 12, 24, and 48 h under the microscope in DAPI (4′,6-diamidino-2-phenylindole) staining. Moreover, PPE activated the apoptosis pathway by increasing the protein expression of cleaved caspase-3. In addition, it also expressed elevation of pro-apoptotic Bax and degradation of anti-apoptotic Bcl-2. In addition, it enhanced the p-Chk2 protein in arresting the cell cycle and increased the protein expression of p-Histone H2A.X. Those results suggest that PPE can reduce cell viability in Caco-2 cells through ROS production and DNA damage.
Keywords: Pomelo peel; Colorectal cancer; Apoptotic cell death; ROS; DNA damage
| 1. Introduction | ▴Top |
Colorectal cancer (CRC) is the most common malignacyt in humans worldwide. It is related to dietary habits, lifestyle, and gene factors, which are critical modulators of the development of human CRC (Ramos et al., 2011). However, most colorectal cancer patients have presented drug resistance, and only 10% can survive after the five-year therapy (Laka et al., 2021). Therefore, developing new natural products for colorectal and other types of cancer and cancer is of high demand.
Pomelo (Citrus grandis) belongs to Rutacea family, a member of citrus fruits and grows in Vietnam, Taiwan, and Thailand. Pomelo peel is a by-product that is ignored and pollutes the environment. Pomelo citrus peel is rich in biologically active polyphenols, especially phenolic acids, and flavonoids, with potential antioxidant, anti-inflammatory, antiproliferative, anti-allergic, anticancer, and antimicrobial functions (Visakh et al., 2022). It is also of interest for application in cosmetic, functional food, and as a natural addative. In addition, these compounds are effective in inhibiting key enzymes in mitochondrial respiration, protecting from heart disease,inflammation and cancer (Anh et al., 2021; Xi et al., 2021). Thus, it is necessary to utilize pomelo peel by-products for pharmaceutical applications. This study aimed to evaluate the anti-proliferation machnism of Citrus grandis peel alcohol extract on Caco-2 carcinoma colon cells. In addition, our study is the first research to assess the anti-colon cancer ability of this resource.
Programmed cell death is a necessary process in the growth and development of humans. Cell growth and cell death control the balance of cells in the body. Programmed cell death includes apoptosis, necrosis, and autophagy while apoptosis is a type I of programmed cell death and type II is autophagy, necrosis is known as a type III (Wu et al., 2020). These processes activate specific signaling pathways; apoptosis and neurosis are two types of cell death but they have different mechanisms. However, autophagy is a degradation mechanism of cells, although it also can induce cell death. If these three types of cell death are put on an axis, following survival superiority, autophagy, and necrosis will be at the opposite ends (Chen et al., 2018). Furthermore, cell death has been recognized and engulfed by nearby phagocytic cells and digested in the lysosome. This programmed cell death is mediated by several signaling pathways including DNA damage, cellular stress, and immune surveillance (Carneiro and El-Deiry, 2020). Two major programs are intrinsic and extrinsic pathways that occur in psychological and pathological situations. The intrinsic pathway is known as a stress happening in mitochondria because of stimulation of some signals such as non-apoptotic, and apoptotic. The extrinsic pathway occurs when extracellular ligands attach to the extracellular domain (Yu et al., 2017).
The apoptotic pathway is controlled by Bcl-2 family members and results in mitochondrial permeability, releasing pro-apoptotic components (Bax, Bak, and anti-apoptotic protein (Bcl-2, Bcl-XL, etc.) to induce cytochrome c into mitochondria space then forming procaspase-9 and APAF-1 (apoptotic protease activating factor 1) complex known as apoptosome (Wang et al., 2020). In addition, the executioner caspase-3 is recruited to the apoptosome, where the resident caspase-9 activates it. Later on, caspase-9 cleaves and activates the executioner caspase-3/6/7, leading to cell apoptosis. Caspase promotes the typical apoptotic features, including DNA fragmentation and cell death in several tissues (Lv et al., 2021).
Reactive oxygen species (ROS) when produced in an imbalanced way in cells, it can not eliminate toxicity or reconstruct the resultant impairment called “ oxidative stress.” ROS also regulate aging, activate the immune system, and cancer (Bardaweel et al., 2018). In oncology, ROS are known to induce apoptosis, a cancer treatment approach. It makes the mitochondria open because of deficient permeability and leads to the release of cytochrome-c to cytosol, then activates the caspase cascade as caspase-9, caspase-3, and -8 and protein are cleavaged, resulting in cell death (Aggarwal et al., 2019). In our study, we evaluated the action mechanism of pomelo peel extract in Caco-2 colon cancer cells and assessed whether it is suitable for alternative therapy for colon cancer medicine in future.
| 2. Materials and methods | ▴Top |
2.1. Sample preparation
Pomelo was purchased in the Longquan and Neipu traditional markets, stored at 4 °C, and used to procurse the peels. The powdered peels (25 g) were then extracted with 500 mL of 50, 75, and 95% ethanolfor 30 min under ultrasound condition using triplicate samples.After filtering, the filtrate was concentrated by using a rotary evaporator, and the residue was frozen at −20 °C and then placed at −80 °C overnight. Freeze-dried powder was stored at −80 °C until use. From our preliminary data of cell viability, an extract of 75% ethanol was found to afford the best results and hence was used in further experimentation. The stock solution wasto desired concentration before use (Im Ahn et al., 2017). The extraction yield after freeze drying was calculated using the formular below (Gunwantrao et al., 2016).
The fresh peel of pomelo was peeled off by hand, washed with tap water to rinse dirt or some insects on the peel, cut into pieces, and dried at 45 °C for 20 h. The dried peel was ground to powder with a blender mortar (HiPoint, Kaohsiung, Taiwan). The powder (25 g) was extracted in 500 mL of 75 % ethanol by ultrasonic for 30 minutes (the sample was triplicated). After filtering (0.45 μm, Merck KGaA, Darmstadt, Germany), the filtrate was concentrated by a rotary evaporator (EYELA, Tokyo, Japan), and the residue was frozen at −20 °C and then placed at −80 °C overnight. Freeze-dried (FD-5030, Panchum,Taiwan) to powder, and powder was stored at −80 degrees until use. The powder was dissolved in 75 % ethanol to a final concentration of 1,000 μg/mL (The stock solution). The stock solution was diluted with free-medium to the desired concentration for our study.
2.2. Chemicals and reagents
The human colon adenocarcinoma Caco-2 cells were purchased from BCRC 60182. In addition, Dulbecco’s Modified Eagle’s Medium (DMEM), Penicillin-Streptomycin, and trypsin were provided by Cytiva, USA, and Fetal Bovine Serum (FBS) was from Hyclone, USA. Gel electrophoresis system, Dye Reagent, SDS Solution, Stacking Gel Buffer, Resolving Gel Buffer, 10X Tris/Glycine/SDS Buffer, 10X Tris/Glycine Buffer, LaemmiLi Sample Buffer, 2-mercaptoethanol, and TEMED were purchased by BIO-RAD, USA. PRO-PREP (Protein et al.) was from iNtRON BIOTECHNOLOGY, Korea. Primary antibody of Caspase-3, -8, -9, Bax, Bcl-2, Bcl-XL β-actin, LC3A/B, p-p53, p-Chk2 and p-Histone H2A.X were from Cell Signalling Technology, USA. Secondary antibody HRP Goat anti-mouse IgG HRP Donkey anti-rabbit IgG were from Biolegand, USA. PVDF membranes were from Millipore, Billerica, MA. Trypan blue solution, dimethyl sulfoxide (DMSO), thiazol blue tetrazolium bromide 98 %: MTT, and other chemicals were from Sigma-Aldrich, USA.
2.3. Cell culture
Caco-2 cells (BCRC 60182) was obtained from the Bioresource Collection and Research Center, Hsinchu, Taiwan, Taiwan) were cultured in DMEM medium are from HyClone Laboratories, Inc. (Logan, UT, USA), suplemented with 10 % Fetal Bovine Serum (FBS) and 1 % penicillin at 37 °C, and humidified 5 % CO2 incubation. The media were changed after two days, and cells were viewed under a phase-contrast microscope (Dino-Lite, Hsinchu, Taiwan, Taiwan) at 200× magnification. The subculture was done when the cells reached 80 % confluence.
2.4. Cell viability and morphology
Cell viability was determined by MTT assay, removing the medium in the cell dish, adding 2 mL PBS to wash the cell, and remove the PBS. Continually 1 mL trypsin was added and incubated at 37 °C for 4 min. Cells were washed by 2 mL DMEM medium into a centrifuge tube and then centrifuged at 1,500 rpm (3.5 g) for 4 min. The supernatant was then and 1 mL medium ws then added followed by homogenization. Ten microliters of suspension cells was taken and mixed with 10 μL of trypan blue into hemocytometer (1:1), count cells under a microscope. In a 96-well plate, to each well was added 100 μL containing 1x104 cells, incubated for 24 or 48 h, removed the medium, and the sample was added following desired time. Afterwards, cells were added with MTT for incubating 4 h, after which 100 μL DMSO were added and shaken at 100 rpm for 30 min. Finally, the plate was read by using an ELISA reader at 540 nm (Shang et al., 2018).
2.5. DNA condensation assay
DAPI (4,6-diamidino-2-phenylindole) staining was one of technology for analyzing DNA condensation. Cells were cultured in a culture dish for 24 h, then incubated with IC50 concentration of PPE ( 455 μg/mL ). The medium was removed and washed three times with PBS, fixed with −20 °C methanol or 4 % paraformaldehyde, for 15 min in 4 °C. Afterward, wash three times with PBS and add 0.1 % Triton X-100 in PBS for 15 min (room temperature). Washed three times with PBS, added DAPI (1μg/mL) to cover the cells, and incubated at 37 °C in the dark for 30 min. Finally, the material was washed three times with PBS, then keep in 4 °C. The cells were photographed by using a fluoresence microscope (Dino-Lite, Hsinchu, Taiwan) (Alam et al., 2015).
2.6. Reactive oxygen species (ROS) staining
The ROS production was detected by comparing the control and treated groups using 2′,7′-dichlorofluorescein diacetate (DCFDA). The dose of IC50 was contacted with cells for a desired time, and wash with DMEM (free serum). After that, 10 μM of DCFDA in DMEM were added and incubated 30 minutes in the dark of the incubator. Then washed three times with PBS and photographed with fluoresent microscope. The blue filter was used because the excited wavelengh is 485 nm and emission wavelength is 580 nm (Sheikh et al., 2015).
2.7. Western Blotting
Caco-2 cancer cells (1 ×107 cells) seeded in a 10 cm2 dishes were treated with IC50 of PPE for 0, 6, 12, 24, and 48 h. The cells were harvested and lysed with PRO-PREP lysis buffer (iNtRON BIOTECHNOLOGY, Boston, USA). The protein concentrations of the cell lysate and subcellular fraction were quantitated with a protein assay kit (Bio-Rad Laboratories, Hercules, California, USA). The total proteins (28 μg) were used for Western blotting analysis, and all samples were used for loading to 10% sodium dodecyl sulfate−polyacrylamide gel electrophoresis (SDS−PAGE) for 100–120 min (Juan et al., 2018). The proteins were then transferred to poly(vinylidene fluoride) (PVDF) membranes (Millipore, Billerica, MA, USA) and incubated with primary antibodies such as LC-3II, Bcl-2, Bcl-xL, Bax, caspase-9, caspase-3, and caspase-8 (Cell Signaling Technology, Danvers, MA, USA), washed three times, and incubated 1–2 hour with horseradish peroxidase (HRP)-conjugated secondary antibody for detection by a ChemiLuciferase kit according to the manufacturer’s instructions (Millipore, Billerica, MA, USA). In addition, we used anti-β-actin (primary antibody) as internal control.
2.8. Statistical analysis
All results from triplicate analyses were shown by the mean ± standard deviation (SD). Then, they were analyzed by one-way analysis of variance (ANOVA) and Duncan’s multiple comparison tests (SAS Institute Inc., Cary, NC, USA) to determine significant differences among treatments, p < 0.05. Finally, all figures were performed by using Sigma Plot version 14.0 (Chu et al., 2013).
| 3. Results and discussion | ▴Top |
To study cancer and to make a plan to treat patients, use of in vitro model is important for understanding the molecular and cellular mechanisms. Elimination of cancer cell viability has been used in the treatment cancer, but to eliminate side effects, applying natural products in the treatment of cancer is considered not only safe but also an inexpensive remedy for patients.
In this study, PPE was obtained by using alcohol in ultrasonic-assisted extration; the extract was tested in Caco-2 cells to check the cell proliferation after 24 h. Concentrations range was 75∼1,000 μg/mL; working doses were diluted in free-medium for testing in the cells. After 24 and 48 h, cell viability and morphology (Figure 1b) strongly suppressed the proliferation of Caco-2 colon carcinoma cells at 75∼1,000 μg/mL in vitro compared to the control group by using MTT (Figure 1a). Cells were reduced by following the concentrations 75, 250, 500, 750, and 1,000 μg/mL. Figure 1a, also revealed IC50 of 24 h treatment at around 483 μg/mL, and IC50 of 48 h treatment approximately at 455 μg/mL, therefore, this study used IC50-455 μg/mL for further experiment.
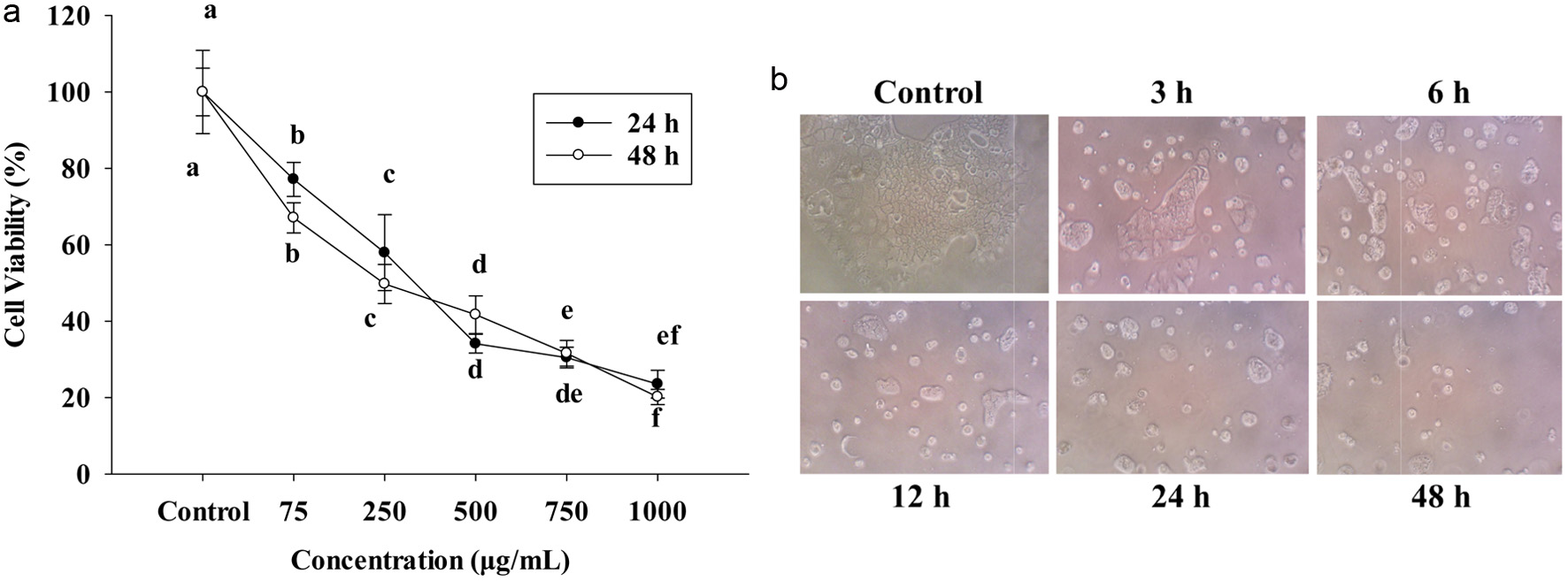 Click for large image | Figure 1. (a) The cell viability at 24 h and 48 h of PPE on Caco-2 cells. Cells were seeded in 96-well plates at 1x104 cells/well and seeded 24 h. 75, 250, 500, 750, 1,000 (μg/mL) PPE incubated in 24 h and 48 h. (b) Cell morphology was treated with IC50 PPE at different times. The cell viability was determined by MTT assay; each value represents mean ± SD (n = 3) (p < 0.05). |
Figure 1b shows the cells treated with or without IC50 of PPE. After the challenge with IC50 455 μg/mL of PPE, the cells were healthy and had more abundant viability at control, 3 and 6 h by observing under an inverted microscope. However, after 12, 24, and 48 h, the number of cell death increased, detached and floated in the medium.
Under normal condition, the content of ROS is low-level and plays a role in a balanced homeostasis that is able to create and eliminate ROS (Wang et al., 2021). However, excess ROS can damage DNA and lipid and cause injury in cells and leads to the generation of carcinogenesis. According to a previous study (Renaudin, 2021), a high level of ROS can decrease tumor growth via activation of cell cycle inhibitor.
On the other hand, 2′,7′-dichlorodihydrofluorescein (DCFDA) staining is a non-fluorescent probe that can pass into the cell membrane, intracellular esterase enzymes will cut the acetate group and combine with DCF. Then fluorescence may be detected by using a fluorescent microscope (Figure 2a). The graphs show a strongly increasing fluorescence in the treatment group after 12, 24, and 48 h (B, C, D), compared with the control group-A. Treated groups had brighter and deeper green light than the untreated control; in untreated control, a dim green color was noted. Thus, ROS penetrates into the cell membrane, reacts, and creates fluorescence, thus increased level of ROS means an increase in apoptosis (Zhang et al., 2023). Figure 2b also expresses the significantly increasing at 12, 24, and 48 h compared to the control group.
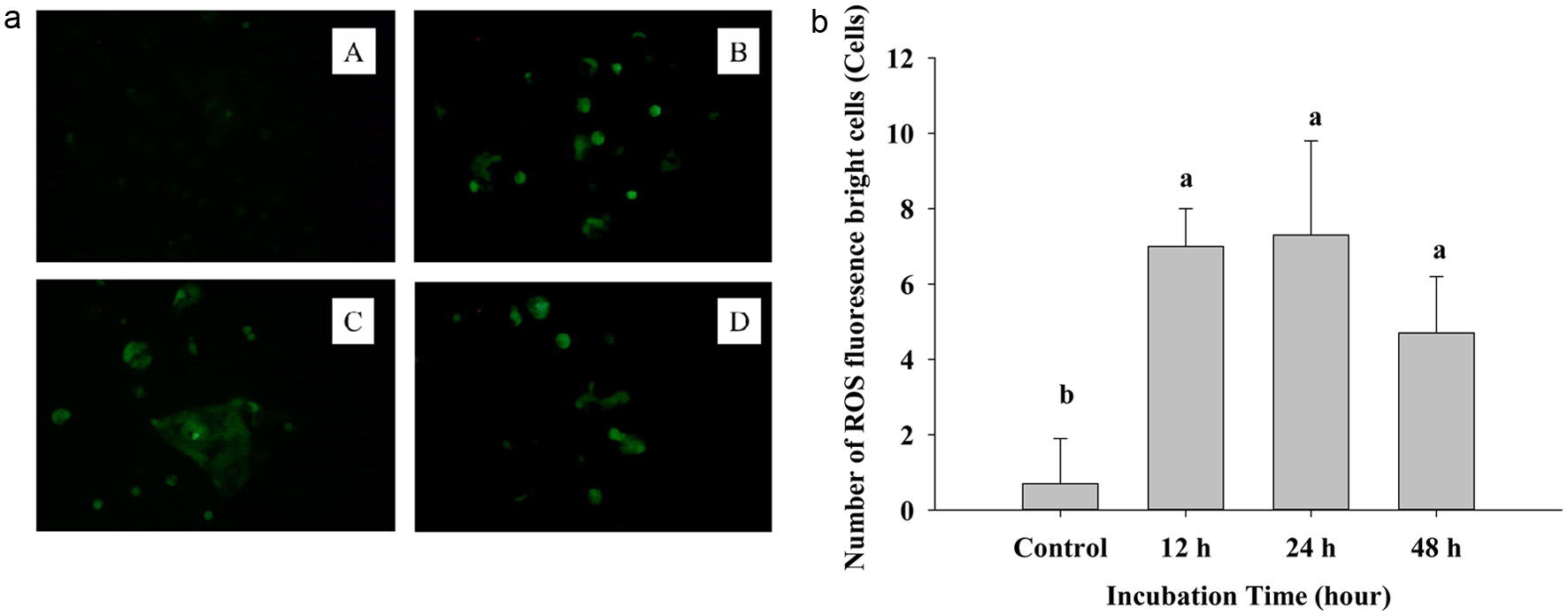 Click for large image | Figure 2. (a) The fluorescence image of DCFH-DA staining cells after treating IC50 of PPE on Caco-2 cells. A: control group, B: 12 h, C: 24 h, D: 48 h. (b) Quantization of Reactive oxygen species generating cells. Each value represents mean ± SD (n = 3) (p < 0.05). |
In Figure 3, Caco-2 cell treatment was stained with DAPI dye to see the nuclear-condensed chromatin of cells; after treatment with IC50-455 μg/mL, see very clearly bright cells, which means nuclear condensation at 12, 24, and 48 h compared to the control. After treating IC50 of PPE for 3 and 6 h, there was no reaction with DAPI staining, and the cells were darker than the 12, 24, and 48 h treatment groups. The bright blue cells indicate cell-exposed DNA damage at 12, 24, and 48 h.
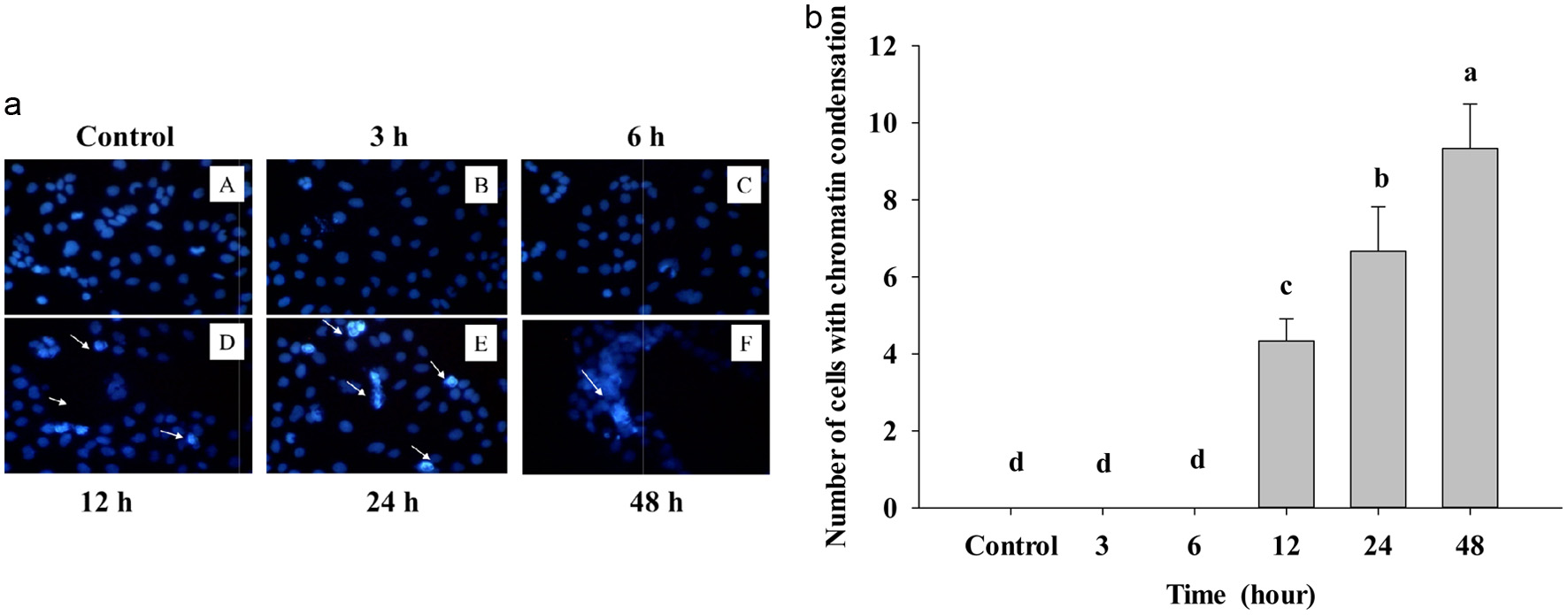 Click for large image | Figure 3. (a) Cells treated with IC50 of PPE at different times and visualized under the fluorescent microscope by DAPI staining. Control – A treated B, C, D, E, and F; the arrow indicated the bright cells. The assay was done after 3, 6, 12, 24, and 48 hours of PPE treatment on Caco-2 cells. (b) Number of chromatin condensation cells per panel by counting in three different dishes. The cells were seeded in the dish and after PPE treatment at other times. Each value represents mean ± SD (n = 3) (p < 0.05). |
It is noteworthy to see cells with strinkage membrane broken, and these changes just show upon apoptosis in cells, after counnting condensed chromatin; as Figure 3b shows that apoptotic cells are significantly increased statistically. Because death cells reveal AT (adenine-thymine) rich region in DNA, DAPI can go through an intact cell membrane, this fluorescent stain strongly binds to DNA, thus resulting in fluorescence of dead cells (white arrow). Actually, DAPI can be used for both live and dead cells, however, staining alive cell presented weak blue light and dead cell explosed to DNA damage showed the bright blue light.
Bcl-2 is for both survival and induced cell death, can lead to cancer growth and prevent cancer treatment. Bax is one member of the B-cell lymphoma (Bcl-2) family, belonging to pro-apoptotic protein and a major pro-apoptotic protein in the Bcl-2 family (El-Sewedy et al., 2023). It is activated by some stimuli from inner cells such as DNA damage, and cytotoxicity. In some research, patients have high Bax levels that can prolong survival if compared to some who have low levels of Bax. For breast cancer patients, deprivation of Bax reduces the drug effect and lower longevity. Bax protein is mediated and takes part in inducing the apoptosis pathway. Under unstimulated conditions, Bax is located in the cytosol of the cell, but apoptosis happens when Bax translocates to interact and insert into mitochonria, resulting cell death (Alam et al., 2022).
To prove the effect of PPE on Caco-2 cancer cells, we used Western Blotting to examine the level of protein expression Bax, Bcl-2, and Bcl-XL. After treatment of PPE, Bax shows a clear time-dependent pattern; it starts to gradually increase protein concentration at 6 to 48 h compared to the control group (Figure 4a and b).
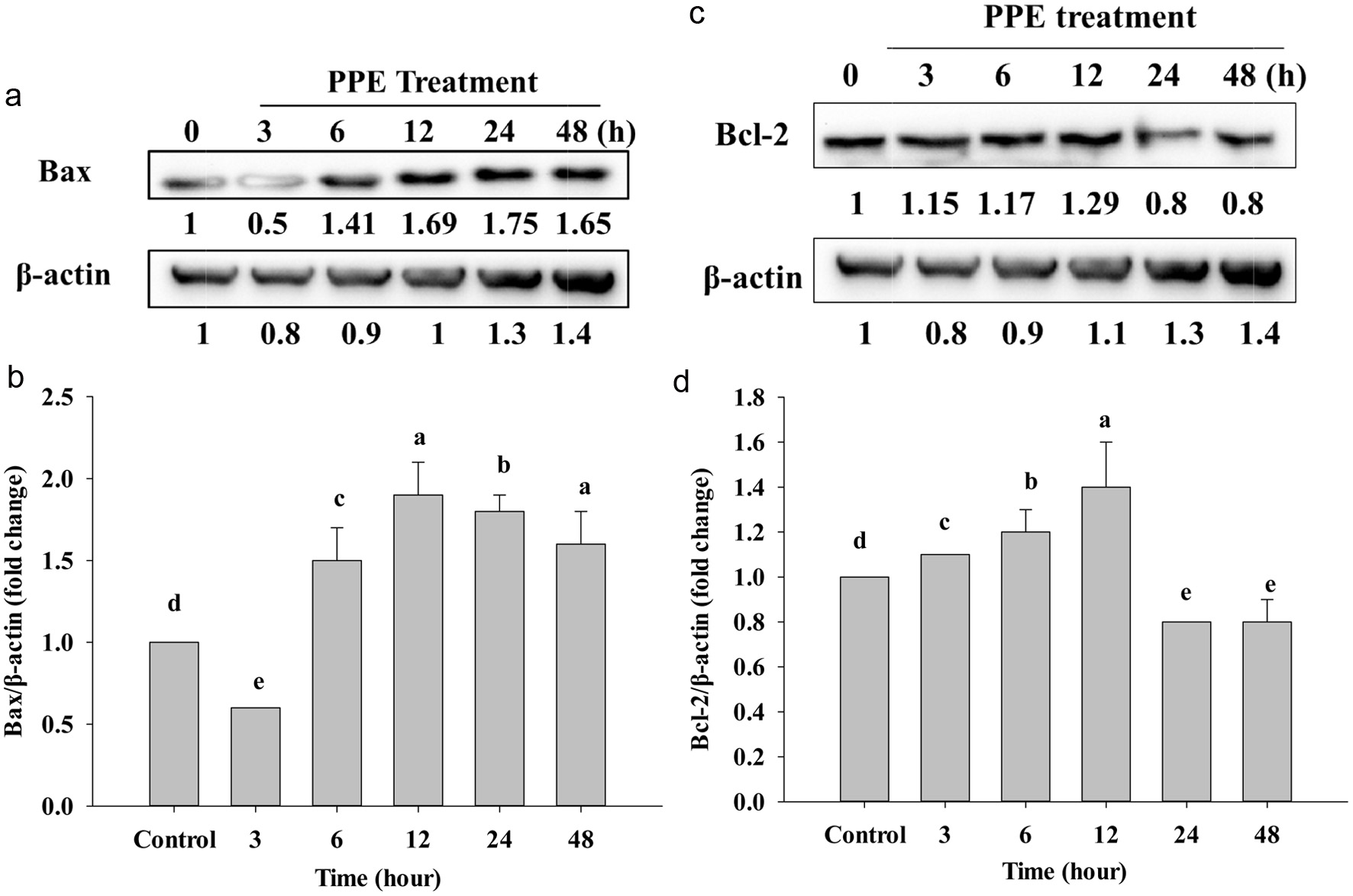 Click for large image | Figure 4. Western Blotting analyzed the protein expression of Bax (a), Bcl-2 (c), and Bcl-xL (e) after treating IC50 of PPE at different times. Quantization result of protein folds change of Bax/β-actin (b), Bcl-2/β-actin (d), and Bcl-xL/β-actin (f). Each value represents mean ± SD (n = 3) (p < 0.05). |
On the other hand, anti-apoptotic Bcl-2 protein also expressed reduction of protein after treatment (Figure 4c). While 3, 6, and 12 h did not significantly decrease, but at 24 and 48 h protein expression significantly decreased compared to the control (Figure 4d). Besides, Figure 4e shows that the level of Bcl-XL protein also suppressed; the band is lighter and lighter at 12, 24, and 48 h; however, at 12, 24, and 48 h protein expression just stated to significantly decrease compared to the control group (Figure 4f). In addition, the increasing of Bax (Figure 4a and b) and decreasing of Bcl-2, Bcl-XL, is due to cell stimulation by PPE, pro-apoptotic Bax could be translocated in the cytosol to enter into mitochondria by making a pore by Bax homo-oligomerization on mitochondrial outer membrane to release cytochrome-c via inhibiting Bcl-2, and Bcl-XL by a combination of BH1, BH2 and BH3 domain to allow cell death (Ritter et al., 2021).
Caspase-3 and caspase-9 are classical hydrolysing enzymes in cancer prevention studies, and they are a member of the caspase family. Caspase-3 was activated through releasing cytochrome c in mitochondria into the cytosol, cytochrome c was combined with Apaf1, caspase-9 was activated by getting a cytochrome c signal, cleaved and activated caspase-3 that exits in the cytosol of cells, was cleaved to be a form of active caspase-3 enzyme, therefore, resulting in apoptosis (Jemmerson et al., 2021). Some research indicate that if cleaved caspase-3 shows high level, it mean that the patient has had an effective cancer treatment such as the head, neck, and breast cancers (Liu et al., 2017). Caspase-9 plays an initiator caspase central role and works with cytochrome c in the cytosol to activate effector caspases like caspase-3, 7, 6. After that, it promote downstream, leading to apoptosis.
In this study, as shown in Figure 5e, after treatment of PPE IC50 on Caco-2 cells, the procaspase-9 band was gradually decreased; Figure 5f shows a 3 to 48 h that decrease was noted compared to the control group. Procaspase-3 will be decreased to cleave to be cleaved caspase-3 that also showed the expression level upregulated after cleavage compared to the control (Figure 5a and b). After activation of Bax, Bcl-2, and Bcl-XL, mitochonrial membrane is broken and releases cytochrome-c, then activated caspase-9 would induce cleavage caspase-3, resulting in cell death. Moreover, procaspase-8 is combined with FADD death domain, and then autocleavage activated, hence cleaved caspase-8 is increased compared to procaspase-8. However, the protein expression of cleaved caspase-8 gradually decreased compared to the control group (Figure 5c and d). The presence of bifunctional apoptosis regulator protein (BAR) can bind to the active caspase-8, thus forming ubiquitination and proteasome-dependent degradation. Therefore, cleaved caspase-8 is decreased compared to the control, thus PPE treatment does notgo through the caspase-8 signaling pathway in this study.
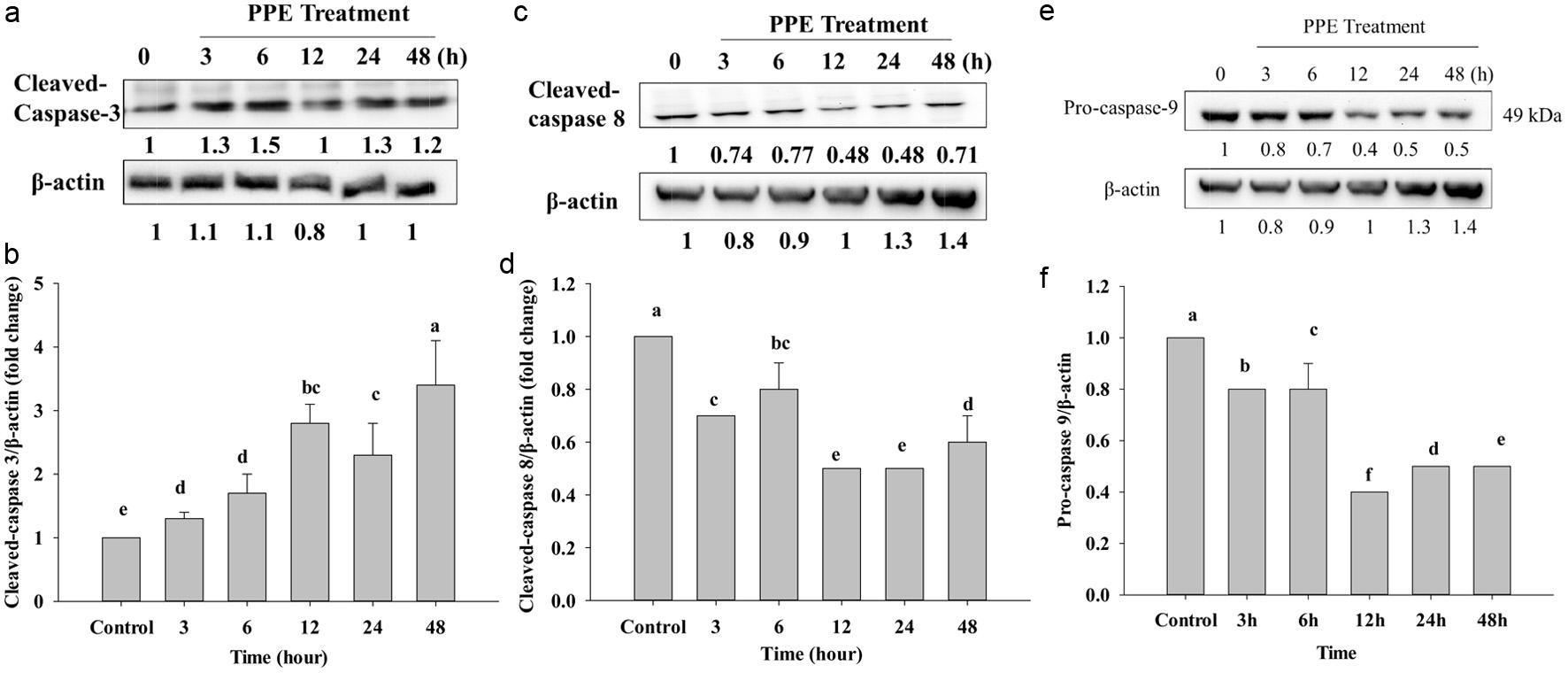 Click for large image | Figure 5. Protein expression of cleaved-caspase-3 (a), cleaved-caspase-8 (c), and pro-caspase-9 (e) after IC50 of PPE treatment, which Western Blotting analyzed. Quantization of protein folds change of cleaved-caspase-3/β-actin (b), cleaved-caspase-8/β-actin (d), and pro-caspase-9/β-actin (f). Each value represents mean ± SD (n = 3) (p < 0.05). |
In cancer, cell cycle arrest is important to stop the cancer cell division, if checkpoint deficiency occurs to be genomic instability and cancer predisposition. Chk2 protein kinase is a protein to checkpoint DNA double strained-break (DSBs), and it is also responsible for DNA repair and through phosphorylating and manage tumor suppressor breast cancer 1 (Brca1) (Hu et al., 2017). Other studies have shown high expression of Chk2, that is a good response indicator for chemotherapy (Gutierrez-Gonzalez et al., 2013).
Chk2 protein kinase works between G1-S and G2-M phase arrest when the G1 and S phases are coupled with ATM; it will allow automatic Chk2 phosphorylation, leading to cell cycle arrest.
The residue of its phosphorylation restrained the transition to the S/G2 phase (Beishline and Azizkhan-Clifford, 2014; Chuang et al., 2023). Figures 6a and b reveal that the treatment of PPE has an effect at 3 to 48 h, and it significantly increased the protein expression of p-Chk2 in time-dependent manners. It might also be due to the PPE-induced cell cycle and p-Chk2-activated DNA damage. Therefore, the cell death of Caco-2 cells might be related to S/G2 phase cell cycle arrest and DNA damage.
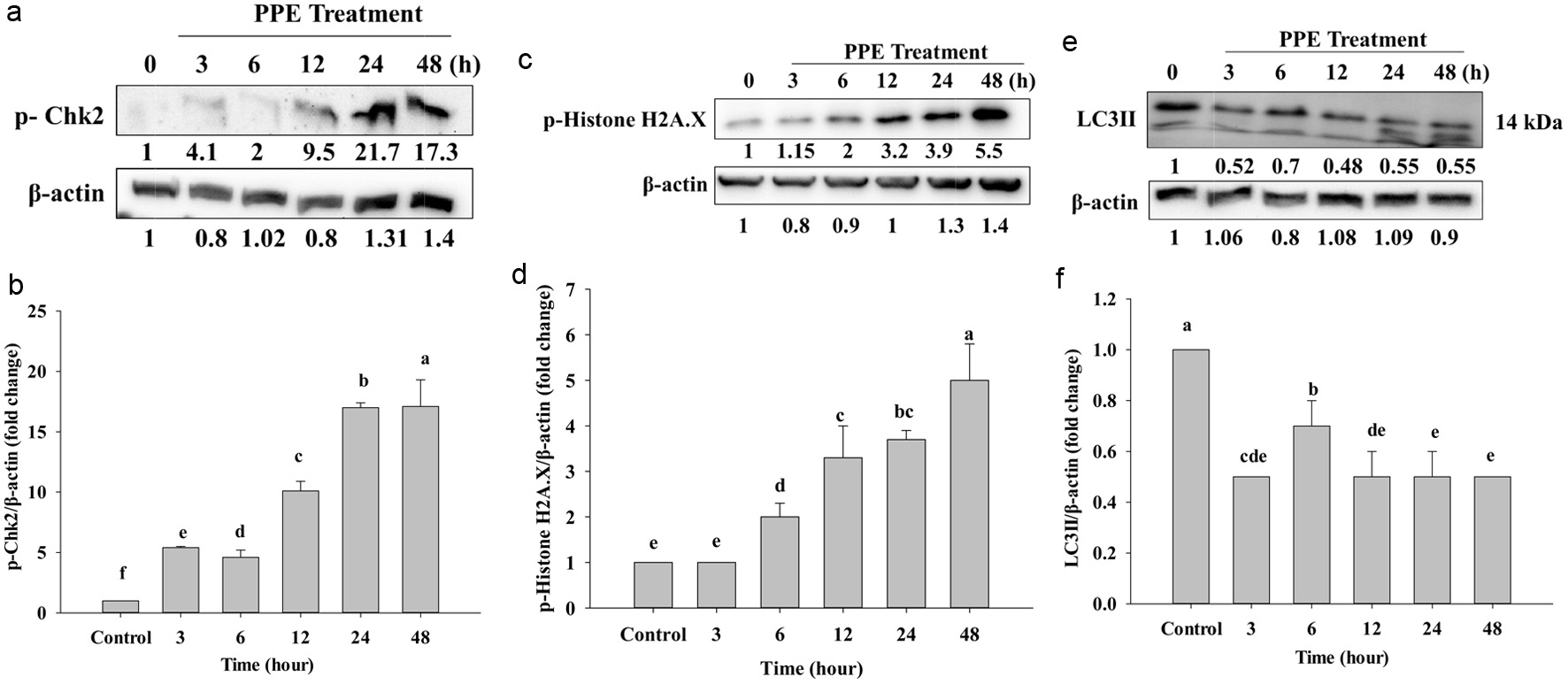 Click for large image | Figure 6. Protein expression of p-Chk2 (a), p-Histone H2A.X (c), and LC3II (e), which Western Blotting analyzed. Quantization of protein folds changes of p-Chk2/β-actin (b), p-Histone H2A.X/β-actin (d), and LC3II/β-actin (f) after IC50 of PPE treatment. Each value represents mean ± SD (n = 3) (p < 0.05). |
Repair of the DNA damage is very important; DNA double-strain-breaks (DSBs) are of high toxicity injury and can lead to cancer, mutation, and chromosomal translocations. In this case, p-Histone.H2AX is responsible for recruiting DNA-damage-response protein to respond to the region gets chromatin damage (Machitani et al., 2020). When DNA is damaged, cells activate DNA damage response (DDR) to recognize and repair. P-Histone.H2AX is a key to repairing DSBs, it transfers signal to start repairing DSBs (Oh et al., 2011). It attracts repair factors around DSBs, and resulting in a high concentration of repair factors. In Figures 6c and d, protein expression of p-Histone H2A.X gradually increased from 6 to 48 h, and the protein concentration increased in a time-dependent manner. At 12, 24, and 48 h, the protein showed deeper and higher protein concentration compared to the control, but not following significantly and in a time-dependent manner as in Figure 6d. That might be because at 12, 24, and 48 h of cells were damaged DNA and the histone H2AX located around DSBs that was phosphorylated to be p-Histone. H2AX it is a major DNA damage response needed to repair the place where has chromatin damage. That is why the protein levels at 12, 24, and 48 h increased.
Both apoptosis and autophagy are involved in programed cell death (Foroozan-Boroojeni et al., 2021). Our results revealed that the process proceeds through the apoptosis pathway. However, wedouble checked on the autophagy pathway as well to find out whether our PPE can result in autophagy; thus western blot was used to detect LC3II. Figures 6e and f show the expression of LC3II on Caco-2 cells; LC3II was strongly decreased following incubation of PPE. However, it did not significantly decrease (Figure 6f), but following 3, 6, 12, 24, and 48 h it totally decreased compared to the control group. The control group had a higher protein than the other groups because the cell was not provided serum when incubated; it is probably amassed, therefore control has a higher concentration than the others. Taken together, PPE might not occur via the autophagy pathway (Figure 7).
Click for large image | Figure 7. The mechanism of PPE-induced apoptosis in human colon cancer Caco-2 cells. |
| 4. Conclusion | ▴Top |
The work presented here demonstrated that pomelo peel extract (PPE) could induce apoptotic death but not autophagy death in Caco-2 cells. Hence, PPE has great potential to be used as an alternative anti-cancer food supplement and functional food ingredient. The world also make good use of the by-products witggood potential for anti-colon cancer alternative therapy in future.
Acknowledgments
This study was funded by 111-2637-B-020-008-. We sincerely appreciate the support from the National Science and Technology Council, and the National Pingtung University of Science and Technology towards this research.
| References | ▴Top |